Today we have another guest post. This one is by Ryan Tibshirani an Assistant Professor in my department. (You might also want to check out the course on Optimization that Ryan teaches jointly with Geoff Gordon.)
Screening and False Discovery Rates
by Ryan Tibshirani
Two years ago, as a TA for Emmanuel Candes’ Theory of Statistics course at Stanford University, I posed a question about screening and false discovery rates on a class homework. Last year, for my undergraduate Data Mining course at CMU, I re-used the same question. The question generated some interesting discussion among the students, so I thought it would be fun to share the idea here. Depending on just how popular Larry’s blog is (or becomes), I may not be able to use it again for this year’s Data Mining course! The question is inspired by conversations at the Hastie-Tibs-Taylor group meetings at Stanford.
Consider a two class problem, genetics inspired, with 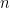
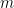
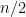


Suppose that we compute a two sample 
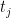
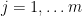
![\displaystyle \mathrm{FDR} = \mathop{\mathbb E}\left[\frac{number\ of\ null\ genes\ called\ significant}{number\ of\ genes\ called\ significant}\right].](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle++%5Cmathrm%7BFDR%7D+%3D+%5Cmathop%7B%5Cmathbb+E%7D%5Cleft%5B%5Cfrac%7Bnumber%5C++of%5C++null%5C++genes%5C++called%5C++significant%7D%7Bnumber%5C+of%5C++genes%5C++called%5C++significant%7D%5Cright%5D.+&bg=ffffff&fg=000000&s=0&c=20201002)
The Benjamini-Hochberg (BH) procedure provides a way to do this, which is best explained using the 





and call the 


An alternative procedure to estimate the FDR uses a null distribution generated by permutations. This means scrambling all of the group labels (healthy/sick) uniformly at random, and then recomputing the 

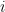
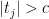


How good are these estimates? To answer this, we’ll look at a simulated example, in which we know the true FDR. Here we have 



Now what happens if, before computing these estimates, we restricted our attention to a small group of genes that looked promising in the first place? Specifically, suppose that we screened for genes based on high between-group variance (between the healthy and sick groups). The idea is to only consider the genes for which there appears to a difference between the healthy and sick groups. Turning to our simluated example, we kept only 1000 of the 2000 genes with the highest between-group variance, and then computed the BH and permutation estimates as usual (as if we were given this screened set to begin with). The plot below shows that the FDR estimates are now quite bad, as they’re way too optimistic.

Here is the interesting part: if we screen by total variance (the variance of all gene expression measurements, pooling the healthy and sick groups), then this problem goes away. The logic behind screening by total variance is that, if there’s not much variability overall, then there’s probably no interesting difference between the healthy and sick groups. In our simulated example, we kept only 1000 of the 2000 genes with the highest total variance, and computed the BH and permutation estimates as usual. We can see below that both estimates of the FDR are actually pretty much as accurate as they were in the first place (with no screening performed), if not a little more conservative.

Why do you think that the estimates after screening by between-group variance and by total variance exhibit such different behaviors? I.e., why is it OK to screen by total variance but not by between-group variance? I’ll share my own thoughts in a future post.

7 Comments
The theory behind most standard statistical procedures is based on the assumption that no data-based pre-selection is done. But quite often such procedures are in fact applied after some pre-selection. There are other examples for this, for example performing a test based on a certain distributional assumption only after having checked the assumption.
Now when is pre-selection harmless, and when isn’t it? Generally I’d think that pre-selection is the less harmful the closer to independent it is to what is actually done in the final statistical procedure. Theory tells us the distribution P(T) of a statistic T. Pre-selection changes this to P(T|S), S meaning that the data were pre-selected. Under independence, P(T)=P(T|S), which means that the original theory for T holds, whereas if S is strongly connected to T, P(T|S) may be quite different from P(T)
Now screening for between-group variance will systematically select genes that will later produce low p-values, because there are strong observed differences between groups. The p-values are our T, so here T depends strongly on S.
The total variance, however, doesn’t say much about mean differences between groups, so S is closer to independence of T, and therefore P(T|S) will be closer to P(T).
The fun thing here seems to be that the total variance in fact tells us something about the group differences because according to the way the data were generated, null genes have a lower total variance. The t-test seems to be blind to this, probably because variance is standardized out of the statistic.
I realize that I’m not 100% confident of what I’m saying because I’d still expect some dependence between S (total variance selection) and T here in the given setup, but much weaker than before.
One could perhaps say, that the first selection rule systematically selects genes with low p-values, because the t-test is driven by a mean difference which is directly related to between-groups variance, whereas the second one selects genes that look promising for reasons that don’t have to do with what the t-test does. If they still have systematically lower p-values, this is not because the selection has biased the t-test, but rather because the selection has used some information that the t-test ignores.
That was a nice problem. I think Christian is exactly right (so I will repeat what he said :)). When you condition on the between-group variance you are just conditioning on the numerator of the t-statistic since (up to a scaling factor) when there are n/2 people in each group, the between-group variance is the same as the square of the difference between the means, i.e. the numerator of the square of the t-statistic (equivalently its absolute value). So you are just sub-selecting features with a low p-value in the screening.
Screening on the total variance on the other hand does not affect anything because the t-statistic standardizes out the total variance and so intuitively is independent of it. I think the more formal way of saying this is that the t-statistic is ancillary and the sample total variance is sufficient for the population variance so they are independent.
I think it depends on how you view the selecting / conditioning. Either it’s pre-selection relative to the “test”, or I would say it’s post-selection relative to the causal effect you are interested in.
I.e., for a “null gene” the gene expression should be statistically (and causally) independent of the health/sickness variable – the t test checks for this (for a linear Gaussian model of dependence).
But the between-group variance is a function of both the gene expression and health, so if you condition on that you’ve created your own variant of Berkson’s paradox. Another way to do it would be let property A = a person is healthy and property B = that person has positive gene expression (for some given null gene). Then clearly P(A|B) = P(A) — the two are independent. But if you reduce you’re data to only those points where at least one of A or B is present, then the sick people in your “conditioned” data set will all have negative gene expression for that gene, introducing a correlation (also, the conditioned population will fairly obviously have higher between group variance).
In terms of causal DAGs (I wonder if this will look right at all in a comment, oh well), I think a null gene should look something like
Gene expression Healthy/Sick
| \ /
| \ /
| \ /
| v v
| Between group variance
v
Total variance
Perhaps ??
Ok, you can argue about whether the variances are “caused” by the two factors, but in the sense that they are a mathematical operations on the data (well, statistics). It’s a general truth that conditioning on a “collider” is going to introduce spurious correlations.
It’s a nice example too.
Ah, sorry, should have been more careful before posting. Clearly in my example the sick people in the conditioned data set would not have property A and therefore *must* have property B (positive gene expression) rather than the otherway round as I said. Also clearly my attempt at drawing a DAG was misguided, it should be Total variance Between group variance <- Healthy/Sick.
Eesh, sorry, DAG: http://pastebin.com/AB8wvw6Z
I agree with Christian. The ‘problem’ here is that the t-test seems to be the ‘wrong’ test to use, as it’s p-values say nothing about a gene being null or non-null since the mean expression for null and non-null is the same.
The variance of the null genes is 1. The variance of the non-null genes is 1.5.
So a procedure selecting for high variance will enrich for non-null genes and low p-values, although like is said above, in a weaker manner than
a procedure selecting for high between-group variance. Imagine that instead of 1 and -1, the expression for sick patients for non-null genes would
have +1000 or -1000 with equal probability as mean (and still st.d. of 1). A procedure selecting for high variance will also select for the non-null genes.
But it will not select for low p-values.
Couldn’t the statistical properties be illustrated without regard to FDR? The bogosity of naively using tests after pre-screens that are not independent of their test statistics is fairly well-known; qq plots of either the chi-squared statistics or log p-values (from the nulls) would illustrate this cleanly – scatterplots of the statistics used in pre-screen and the main test would illustrate dependence.
Using FDR as above is of course not wrong, but one ends up illustrating the (mild) conservatism of BH, and having to address issues of precisely which flavor of FDR one is estimating. Neither seems strongly related to the main point being illustrated.